








Blog - 01/09/2023
Come trattare l’artrite agendo sull’infiammazione e gestendo il dolore del paziente con risultati efficaci: questo il tema dell’edizione 2023 del World PhysioTherapy Day (8 settembre).

Blog - 21/06/2023
Il valore di un’azienda è frutto di un’armonia di fattori che, mettendo in relazione persone e prodotti, generano circoli virtuosi da cui emergono il talento di ogni risorsa e l’efficacia del...
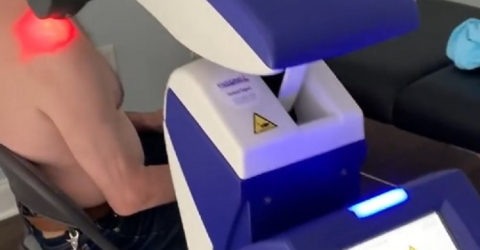
Blog - 21/05/2020
In epoca di Covid-19 c’è un nuovo termine che è entrato a far parte del vocabolario di ciascuno di noi e ha portato ad una sostanziale revisione dello stile di vita e di lavoro: “distanziamento...

News - 11/12/2024
Che si tratti di partecipare a Congressi e Fiere, di organizzare corsi on site o webinar, ASA, anche nel 2024, si è dimostrata fortemente proattiva sia in ambito umano che veterinario.

News - 09/12/2024
Ricerca e Sviluppo sono i pilastri su cui tutte le aziende del Gruppo El.En. fondano la propria crescita: innovazione tecnologica, formazione continua e condivisione di know-how le parole chiave che...

News - 28/11/2024
Due giorni di approfondimenti e seminari focalizzati su Laserterapia MLS® con il supporto di luminari della laserterapia.